
The content in this section seeks to provide a clinical summary of information and resources for medical representatives treating 22q patients.
Clinical Summary
The diagnosis of the 22q11.2 deletion syndrome is suspected in individuals with a range of findings that may include some combination of the following:
- Congenital heart disease (particularly conotruncal malformations) don’t think parents will know what this is maybe go with most frequent as a list (such as: tetralogy of Fallot, VSD, interrupted aortic arch, truncus arteriosus, vascular ring, ASD)
- Palatal abnormalities (likewise –would suggest for example cleft plate, cleft lip and palate, bifid uvula or most often velopharyngeal insufficiency/dysfunction (VPI/VPD)
- Trouble fighting infection (Immune deficiency)
- Low calcium (Hypocalcemia)
- Feeding and swallowing problems
- Kidney differences (renal agenesis, hydronephrosis, multicystic/dysplastic kidneys, duplicated kidney, horseshoe kidney)
- Learning difficulties
- Differences in attention (ADHD/ADD)
- Differences in behavior (anxiety)
Less frequent associations include:
- Growth hormone deficiency
- Autoimmune disease (thrombocytopenia, juvenile rheumatoid arthritis, Grave’s disease, vitiligo, neutropenia, hemolytic anemia)
- Hearing loss (sensorineural and conductive), abnormalities of the inner and outer ear (preauricular pits and tags, microtia, cochlear differences)
- Psychiatric illness
Other structural anomalies:
- Bony differences: differences in the bones of the neck and spine (vertebral differences), extra fingers/toes (pre and postaxial polydactyly of the hands and postaxial polydactyly of the feet), extra ribs, differences in the wing bones (scapula), club foot, premature fusion of the bones of the skull (craniosynostosis)
- Genitourinary tract anomalies: Hernia (umbilical and inguinal) absent uterus, hypospadias, and undescended testes
- Abnormalities of the airway (Laryngotrachealesophageal differences including problems with the windpipe (trachea) and feeding tube (esophagus) including a vascular ring, laryngeal web, trachealesophageal fistula, esophageal atresia, tracheal atresia
- Eye findings (tortuous retinal vessels, ptosis, posterior embryotoxin, scleracornea, coloboma, cataract, and strabismus)
- Differences in the brain and spinal cord (CNS) (cerebellar atrophy, polymicrogyria, enlarged sylvian fissures, neural tube defects, tethered cord, unprovoked seizures, and asymmetric crying facies)
- Gastrointestinal anomalies (GI) (anteriorly placed anus or imperforate anus, esophageal atresia, jejunal atresia, accessory spleens, umbilical hernia, diaphragmatic hernia, intestinal malrotation, and Hirshprung disease
Rare Malignancies:
- Hepatoblastoma, renal cell carcinoma, Wilm’s tumor, neuroblastoma, thyroid carcinoma, melanoma, leukemia
Cytogenetic testing. A small percentage (<1%) of individuals with clinical findings of the 22q11.2 deletion syndrome have chromosomal rearrangements involving 22q11.2, such as a translocation between chromosome 22 and another chromosome.
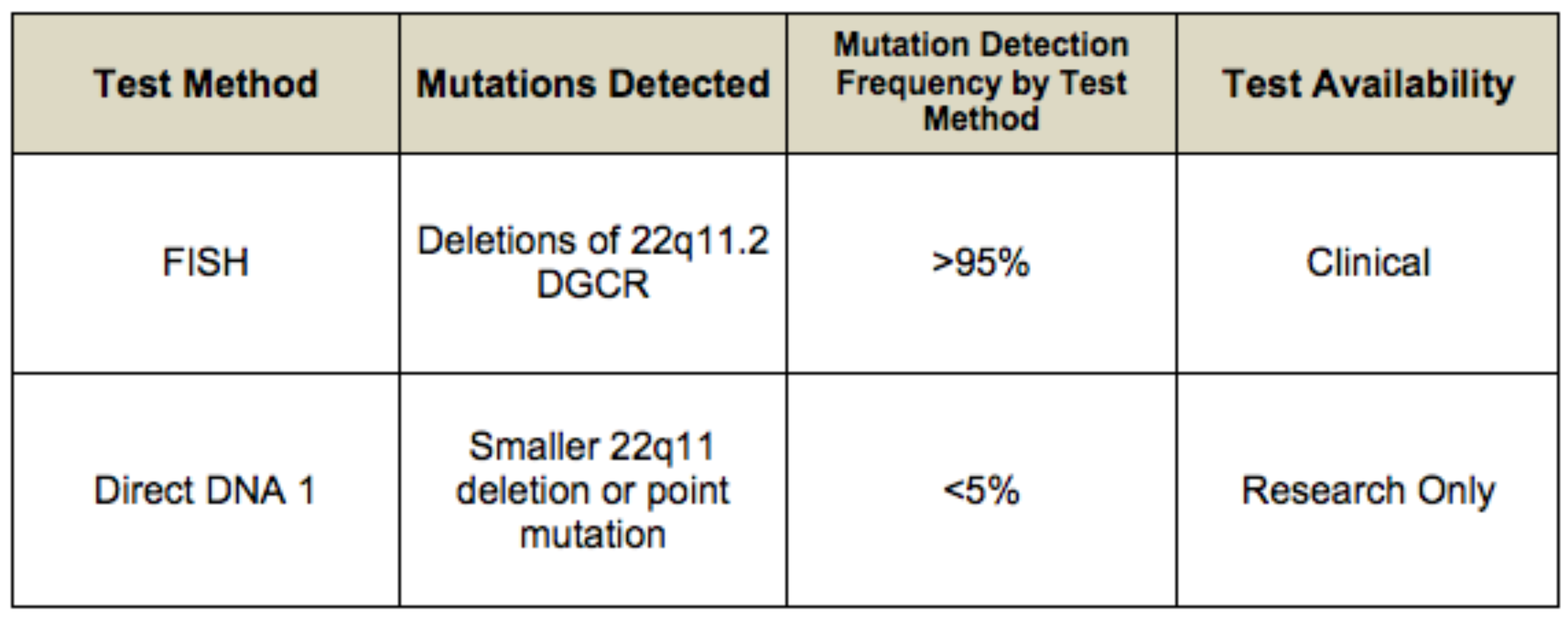
Fish testing will identify ~85% of deletions; the remaining ~15 of deletions require other testing such as MLPA, comparative genomic hybridization (CGH) or SNP microarray testing
Clinical testing
- In 1982, only chromosome studies could identify 22q11.2 deletions by looking at them under the microscope
- In 1991 FISH (fluorescence in situ hybridization) probes were developed that detect ~86% pf deletions
- The two probes commercially available for 22q11.2 FISH analysis are TUPLE1 and N25
- The detection rate of FISH analysis using either probe is thought to be equivalent; however, FISH using either one of these probes is not sensitive enough to detect smaller deletions (<40 kb) within 22q11.2.
- Newer and more precise tests include MLPA (Multiplex ligation-dependent probe amplification), CGH (comparative genomic hybridization), and SNP microarrays
- These tests are useful because they identify small deletions that would be missed by FISH
- They identify 22q11.2 duplications
- CGH and microarrays do not require an elevated index of suspicion of the deletion or duplication
Gene. The 22q11.2 results in ~50 genes missing in the region. [Driscoll et al 1992, Wilson et al 1992, Desmaze et al 1993, Driscoll et al 1993]. This sets up a blueprint for the body to form affecting cells derived from the neural crest often resulting in the birth defects outlined above. Important genes identified thus far in the region include: TBX1, COMT, SCARF2, SNAP29, PRODH, Pl4KA, GPIbβ and CRKL1.
Clinical uses
- Confirmatory diagnostic testing
- Parental testing
- Prenatal diagnosis
Research testing. A few individuals with findings of the 22q11.2 deletion syndrome have normal routine cytogenetic studies and no deletion by FISH, MLPA, CGH or microarray. These individuals may have a change in a gene within the regions, such as TBX1, or a different condition such as CHARGE syndrome.
Table 1. Testing Used in 22q11.2 Deletion Syndrome
When a deletion 22q11.2 is suspected, it is recommended that routine cytogenetic analysis be performed at the time of FISH/MLPA/CGH/array testing because a small percentage (<1%) of individuals with clinical findings of the 22q11.2 deletion syndrome have chromosomal rearrangements involving 22q11.2, such as a translocation between chromosome 22 and another chromosome.
No other phenotypes are associated with deletion of 22q11.2.
Differential Diagnosis
For current information on availability of genetic testing for disorders included in this section, see GeneTests Laboratory Directory. —ED.
Each of the anomalies seen in the 22q11.2 deletion syndrome can be found as an isolated anomaly in an otherwise normal individual.
Up to 8% of individuals with an isolated palatal cleft, including submucosal cleft, may have deletion 22q11.2, making this the most common genetic syndrome associated with palatal clefts. Conversely, the 22q11.2 deletion syndrome is the most common genetic basis of congenital velopharyngeal incompetence.
Disorders with overlapping features:
- Smith-Lemli-Opitz syndrome (when polydactyly and cleft palate are present). Smith-Lemli-Opitz syndrome is associated with elevated serum concentration of 7-dehydrocholesterol (7-DHC) or an elevated 7-dehydrocholesterol:cholesterol ratio. Molecular genetic testing for mutations of the DHCR7 gene is available.
- Alagille syndrome (when butterfly vertebrae, congenital heart disease, and posterior embryotoxon are present). Sequence analysis of the JAG1 gene detects mutations in more than 70% of individuals who meet clinical diagnostic criteria. FISH detects a microdeletion of 20p12, including the entire JAG1 gene, in approximately 5-7% of affected individuals.
- VATER syndrome (when heart disease, vertebral, renal, and limb anomalies are present)
- Oculo-auriculo vertebral (Goldenhar) syndrome (when ear anomalies, vertebral defects, heart disease, renal anomalies are present)
Individuals suspected of having the 22q11.2 deletion syndrome but having normal FISH studies may have a chromosome abnormality involving some other chromosomal region, including deletion 10p13-p14.
Natural History
Findings in 250 individuals (48% male; 52% female) with 22q11.2 deletion syndrome are summarized below [McDonald-McGinn et al 1999b]. In unpublished data on 600 individuals, the percentages for the following findings remain the same [Author, unpublished data 2005].
Thirty-three percent of individuals were five years of age or younger. Marked inter- and intrafamilial variability is observed.
Genotype-Phenotype Correlations
The majority of individuals have the same large deletion of the DGCR. Of note, the size of the deletion remains unchanged with parent-to-child transmission. The great inter- and intrafamilial clinical variability makes genotype-phenotype correlations difficult [Driscoll et al 1995, McDonald-McGinn et al 2001]. Anecdotally, developmental delays appear to be more significant in familial cases; however, this may reflect socioeconomic rather than genetic factors.
Penetrance
Penetrance is complete in individuals with deletion 22q112 detected by FISH; variability is marked.
Anticipation
To date, anticipation has not been observed.
Nomenclature
It is now recognized that the 22q11.2 deletion syndrome encompasses the phenotypes previously called DiGeorge syndrome (DGS), velocardiofacial syndrome (VCFS) (Shprintzen syndrome), conotruncal anomaly face syndrome (CTAF) [Matsuoka et al 1994], many cases of the autosomal dominant Opitz G/BBB syndrome [McDonald-McGinn et al 1995, Fryburg et al 1996, LaCassie & Arriaza 1996], and Cayler cardiofacial syndrome (asymmetric crying facies) [Giannotti et al 1994]. The clinical descriptions of DGS, VCFS, and CTAF resulted from an ascertainment bias.
DGS was originally described as a developmental field defect of the third and fourth pharyngeal pouches with a conotruncal cardiac anomaly and aplasia or hypoplasia of the thymus gland and parathyroid glands. Later, congenital heart disease was added. The majority of individuals with DGS were identified in the neonatal period with a major congenital heart defect, hypocalcemia, and immunodeficiency.
VCFS, also called Shprintzen syndrome, was originally described as the combination of velopharyngeal incompetence (VPI), congenital heart disease (usually a ventricular septal defect or tetralogy of Fallot), characteristic facial features, and developmental delay or learning difficulties. Children with VCFS tended to be diagnosed in cleft palate clinics or craniofacial centers when they reached school age and speech and learning difficulties became evident [Wilson et al 1993, Wulfsberg et al 1996, McDonald-McGinn et al 1997b, Thomas & Graham 1997].
Prevalence
Estimates of prevalence vary from one in 4000 [Wilson et al 1994] to one in 6395 [Devriendt et al 1998]. Given the variable expression of the deletion 22q11.2, the incidence is probably much higher than previously estimated. In a population-based study in Sweden, the mean annual incidence was 14.1 per 100,000 live births [Oskarsdottir et al 2004, Oskarsdottir et al 2005a, Oskarsdottir et al 2005b]. A U.S. population-based study conducted by the Centers for Disease Control (CDC) found an overall prevalence of about one in 6000 in whites, blacks, and Asians, and one in 3800 in Hispanics [Botto et al 2003].